x

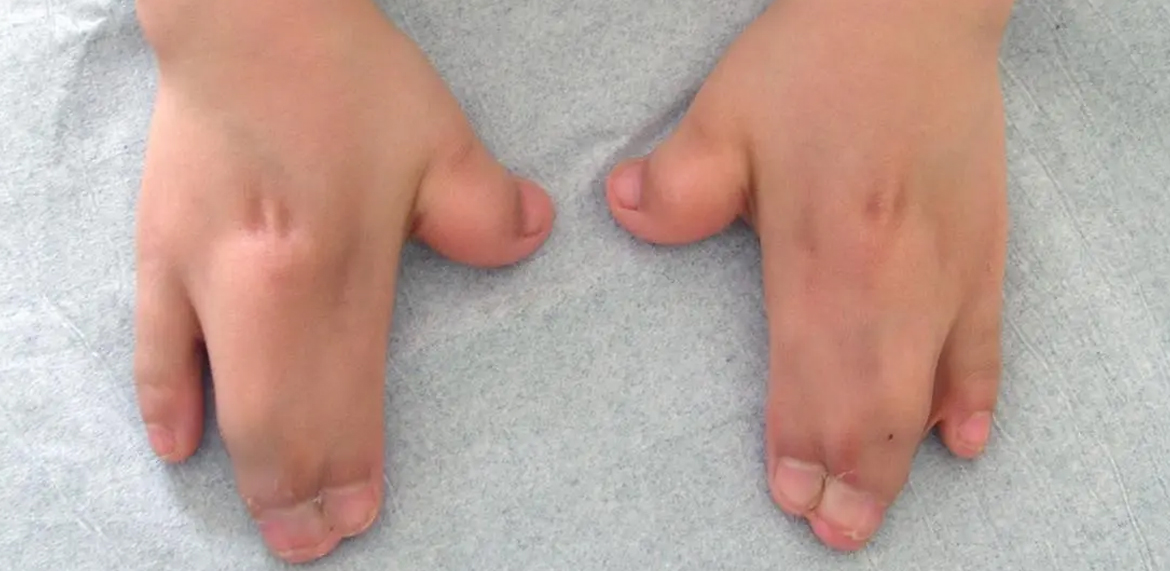
Apert syndrome, also known as acrocephalosyndactyly type I, is a rare genetic disorder characterized by abnormalities affecting the skull, face, hands, and feet. This condition is classified as a type of craniosynostosis syndrome, where one or more of the fibrous joints (sutures) between the bones of the skull close prematurely, leading to abnormal skull shape and facial features.
Key Features of Apert Syndrome:
- Craniofacial Abnormalities: The premature fusion of
cranial sutures leads to a distinctive skull shape, known as acrocephaly
or oxycephaly, characterized by a tower-shaped appearance. This abnormal
skull shape may result in increased pressure within the skull, leading
to potential neurological complications. Additionally, individuals with
Apert syndrome may have characteristic facial features such as:
- Midface retrusion
- Protruding or beaked nose
- Underdeveloped upper jaw (maxillary hypoplasia)
- Wide-set, bulging eyes (hypertelorism)
- Hand and Foot Anomalies: Syndactyly, the fusion of digits, is a hallmark feature of Apert syndrome. Typically, there is fusion of the second, third, and fourth fingers and toes, giving rise to a distinctive appearance known as "mitten hands" and "mitten feet." Additionally, individuals may have other hand and foot anomalies such as short, broad thumbs and toes, and bony fusion of certain joints.
- Dental Abnormalities: Dental issues are common in individuals with Apert syndrome, including overcrowding of teeth, malocclusion (misalignment of the teeth), and delayed tooth eruption.
- Other Manifestations: In addition to the skeletal and craniofacial abnormalities, individuals with Apert syndrome may experience other medical issues such as hearing loss, respiratory problems, and developmental delays.
Causes and Symptoms:
The premature fusion of cranial sutures and the subsequent abnormal skull shape are the primary causes of the physical characteristics associated with Apert syndrome. The specific genetic mutation responsible for Apert syndrome affects the fibroblast growth factor receptor 2 (FGFR2) gene, which plays a crucial role in the development of bones and tissues in the body. This mutation leads to the premature fusion of certain bones in the skull and affects the growth and development of other facial features, hands, and feet.
The characteristic features of Apert syndrome can vary widely among affected individuals, but common symptoms may include:
- Abnormal skull shape (acrocephaly or oxycephaly)
- Midface retrusion
- Protruding or beaked nose
- Syndactyly (fusion of fingers and toes)
- Wide-set, bulging eyes (hypertelorism)
- Underdeveloped upper jaw (maxillary hypoplasia)
- Short, broad thumbs and toes
- Malocclusion (misalignment of the teeth)
- Dental abnormalities (overcrowding, delayed tooth eruption)
- Hearing loss
- Respiratory problems
- Developmental delays
Treatment Strategies:
Management of Apert syndrome typically involves a multidisciplinary approach coordinated by a team of healthcare professionals, including craniofacial surgeons, orthodontists, neurosurgeons, otolaryngologists, and developmental specialists. Treatment may include:
- Surgical intervention to correct craniosynostosis, relieve intracranial pressure, and address facial and hand anomalies.
- Orthodontic treatment to manage dental issues and malocclusion.
- Early intervention and developmental support to address potential developmental delays.
- Ongoing monitoring and management of associated medical complications.
While there is no cure for Apert syndrome, early diagnosis and comprehensive management can significantly improve the quality of life and long-term outcomes for affected individuals.
Prevention:
Since Apert syndrome is a genetic disorder, it cannot be prevented. However, genetic counseling may be beneficial for individuals with a family history of the condition or those planning to have children. Genetic counselors can provide information about the risk of passing the condition to future generations and discuss available options for family planning and prenatal testing.
In conclusion, Apert syndrome is a complex condition that requires multidisciplinary management to address its various manifestations. Early intervention and ongoing care can help improve the quality of life for individuals affected by this rare genetic disorder.